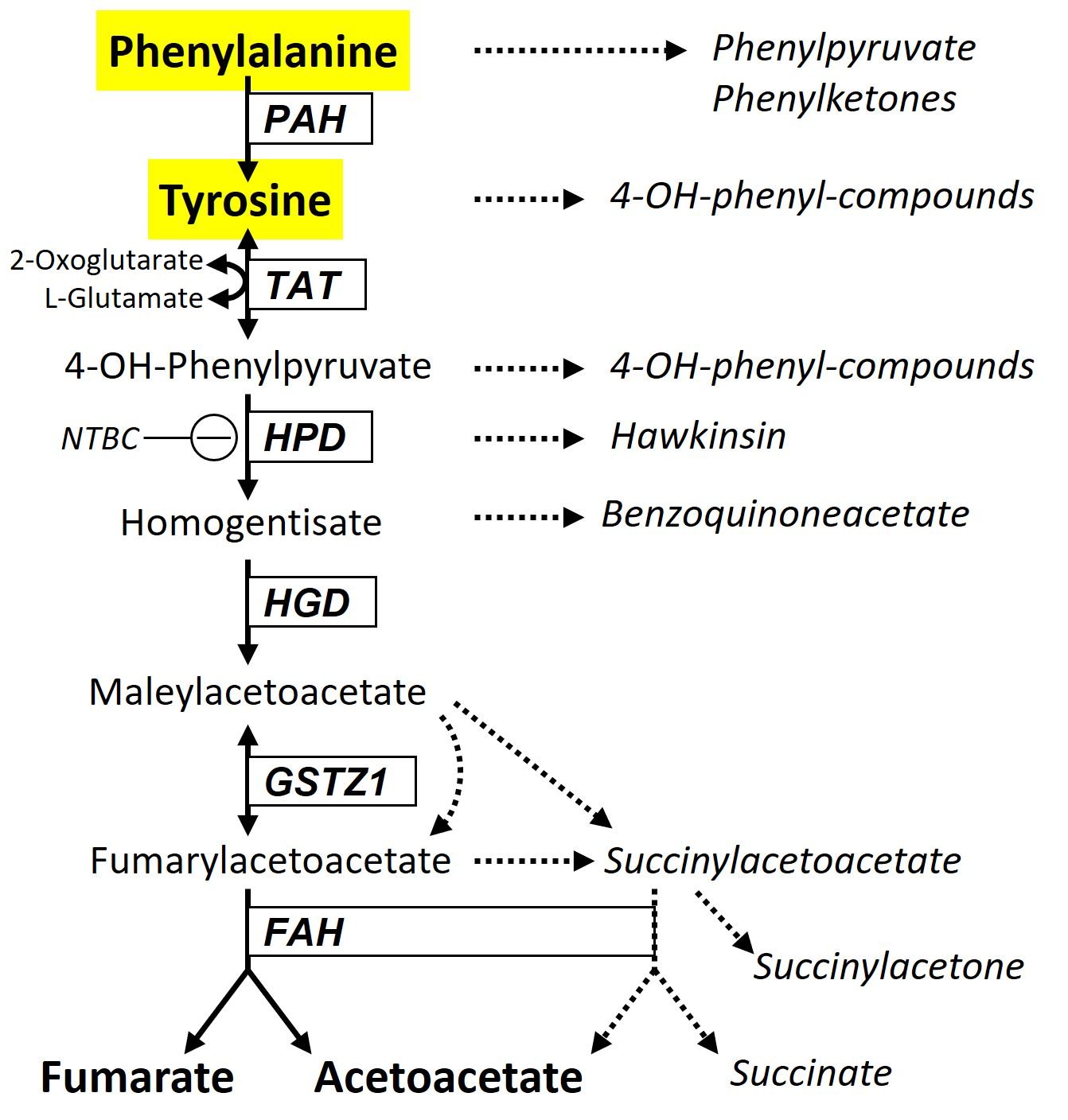
Phenylalanine (Phe) and tyrosine (TYR) metabolism takes place in the cytoplasm.
Breakdown:
- PAH: Phe hydroxylase, requires cofactor tetrahydrobiopterin (BH4); deficiency causes the accumulation of Phe which is transaminated to phenylpyruvate and other phenylketones.
- TAT: TYR aminotransferase = reversible transamination
- HPD: 4-OH-phenylpyruvate dioxygenase; subfunction deficiency by a specific mutation can generate the complex amino acid hawkinsin.
- HGD: homogentisate dioxygenase = cleavage of the phenolic ring is achieved by (HGD).
- GSTZ1: glutathione S-transferase zeta-1 = isomerization of maleylacetoacetate to fumarylacetoacetate, may also happen non-enzymatically.
- FAH: fumarylacetoacetate hydrolase; deficiency causes accumulation of maleylacetoacetate, fumarylacetoacetate, and succinylacetoacetate and ‑acetone. These highly toxic substances inhibit or interfere with several enzymes such as HPD (↑TYR), methionine adenosyltransferase (↑Met), and aminolaevulinate dehydratase (→porphyrias), and are carcinogenic (alkylation of DNA).
Biosynthetic enzymes (not shown) using TYR as precursor: